This shows that there are actually 6 commands containing “rank”; you can now type help() for any of those to get more detail.
If you run the HTML help you will see a heading entitled “Packages”. This will list the packages that you have installed with R. The basic package is ‘base’ and comes with another called ‘stats’. These two form the core of R. If you navigate to one of those you can browse through all the commands available.
R comes with a number of data sets. Many of the help topics come with examples. Usually these involve data sets that are already included. This allows you to copy and paste the commands into the console and see what happens.
R has great graphical power but it is not a point and click interface. This means that you must use typed commands to get it to produce the graphs you desire. This can be a bit tedious at first but once you have the hang of it you can save a list of useful commands as text that you can copy and paste into the R command line.
Bar Charts
The bar chart (or column chart) is a familiar type of graph and a useful graphical tool that may be used in a variety of ways. In essence a bar chart shows the magnitude of items in categories, each bar being a single category (or item). The basic command is barplot() and there are many potential parameters that can be used with it, here are some of the most basic:
x – the data to plot; either a single vector or a matrix.
arg – the names to appear under the bars, if the data has a names attribute this will be used by default.
beside – used in multi-category plots. The default (FALSE) will create a bar for each group of categories as a stack. If you set beside = TRUE the bars will appear separately in blocks.
horiz – the default is for vertical bars (columns), set horiz = TRUE to get horizontal bars. Note however that the bottom axis is always x and the vertical y when it comes to labelling.
col – the colours to use for the bars.
xlab – a text label for the x-axis (the bottom axis, even if horiz = TRUE).
ylab – a text label for the y-axis (the left axis, even if horiz = TRUE).
main – an overall plot title.
legend – should the chart incorporate a legend (the default is FALSE). If you include a legend it defaults to the middle of the right axis.
… – there are many other options!
It is easiest to get to grips with the various options by seeing some examples.
Single category bar charts
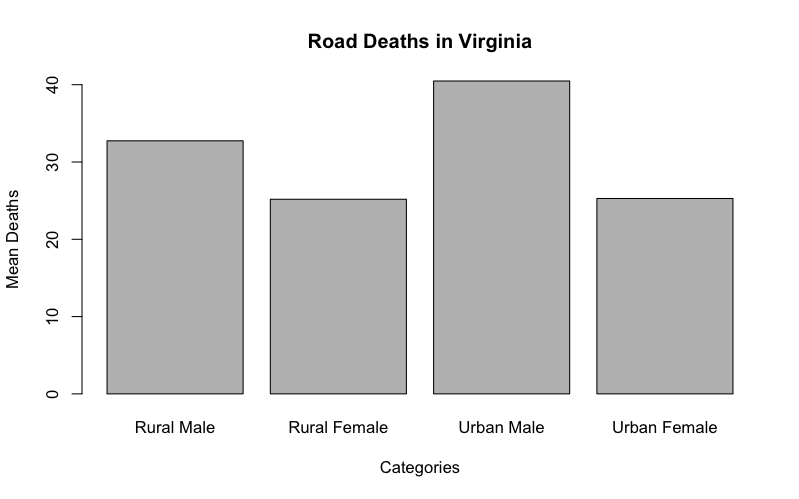
The simplest kind of bar chart is where you have a sample of values like so:
> VADmeans = colMeans(VADeaths)
> VADmeans
Rural Male Rural Female Urban Male Urban Female
32.74 25.18 40.48 25.28
The colMeans() command has produced a single sample of 4 values from the dataset VADeaths (these data are built-in to R). Each value has a name (taken from the columns of the original data). So, you have one row of data split into 4 categories, each will form a bar:
> barplot(VADmeans, main = "Road Deaths in Virginia", xlab = "Categories", ylab = "Mean Deaths")
In this case the bars are labelled with the names from the data but if there were no names, or you wanted different ones, you would need to specify them explicitly:
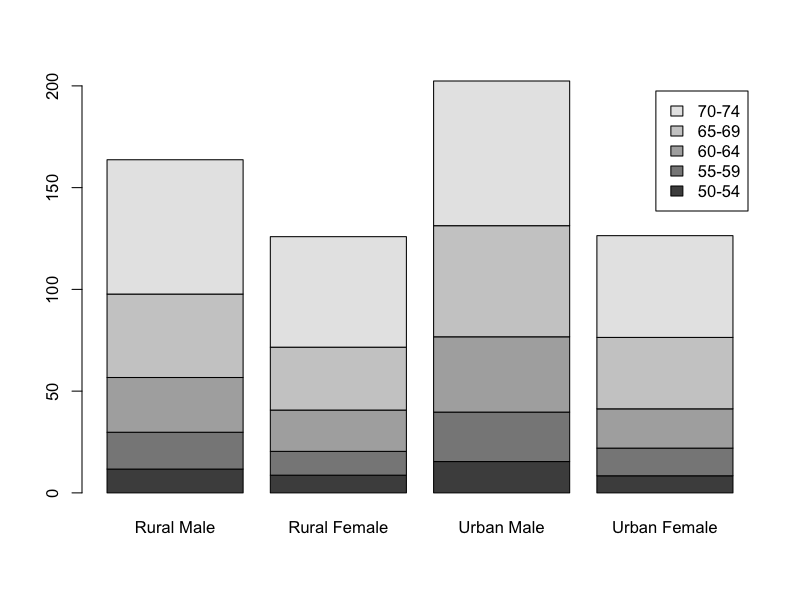
The columns form one set of categories (the gender and location), the rows form another set (the age group). If you create a bar chart the default will be to group the data into columns, split by row (in other words a stacked bar chart).
There are various ways you can present these data.
Stacked bar charts
The default when you have a matrix of values is to present a stacked bar chart where the columns form the main set of bars:
> barplot(VADeaths, legend = TRUE)
Here the legend parameter was added to give an indication of which part of each bar relates to which age group. There are many additional parameters that “tweak” the legend!
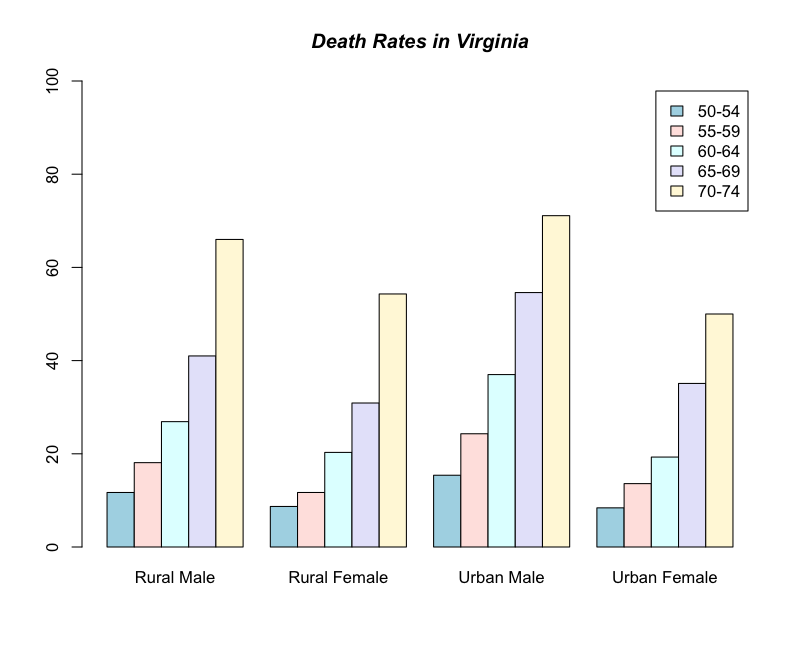
Grouped bar charts
If you want to present the categories entirely separately (i.e. grouped instead of stacked) then you use the beside = TRUE parameter.
> barplot(VADeaths, legend = TRUE, beside = TRUE)
This is fine but the colour scheme is kind of boring. Here is a new set of commands:
This is a bit better. We have specified a list of colours to use for the bars. Note how the list is in the form c(item1, item2, item3, item4). The command ylim sets the limits of the y-axis. In this case a lower limit of 0 and an upper of 100. The command is in the form ylim= c(lower, upper) and note again the use of the c(item1, item2) format. The legend takes the names from the row names of the datafile. The y-axis has been extended to accommodate the legend box.
It is possible to specify the title of the graph as a separate command, which is what was done above. The command title() achieves this but of course it only works when a graphics window is already open. The command font.main sets the typeface, 4 produces bold italic font.
Bar chart of transposed data
The default behavior in the barplot() command is to draw the bars based on the columns. If you wanted to draw the rows instead then you need to transpose the matrix. The t() command will do this. Try the following for yourself:
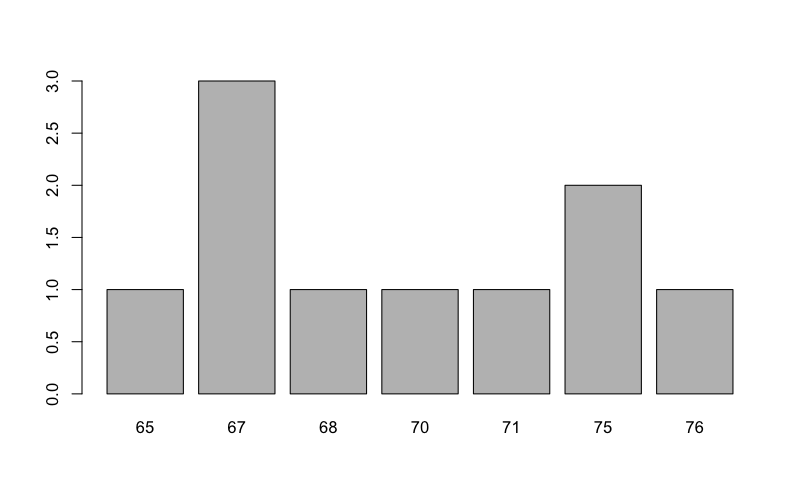
Sometimes you will have a single column of data that you wish to summarize. A common use of a bar chart is to produce a frequency plot showing the number of items in various ranges. Here is a vector of numbers:
Oops. That wasn’t really what you wanted at all. What’s happened is that each item has been plotted as a separate entity. You need to tabulate the frequencies. Fortunately there is an easy way to do this. You use the table() function. Let’s redraw the graph but using the following:
> barplot(table(carb))
This is much better. Now you have the frequencies for the data arranged in several categories (sometimes called bins). As with other graphs you can add titles to axes and to the main graph.
You can look at the table() function directly to see what it produces.
You can see that the function has summarized the data for us into various numerical categories. Note that is not a “proper” histogram (you’ll see these shortly), but it can be useful.
you may wish to show the frequencies as a proportion of the total rather than as raw data. To do this you simply divide each item by the total number of items in your dataset:
barplot(table(carb)/length(carb))
This shows exactly the same pattern but now the total of all the bars add up to one.
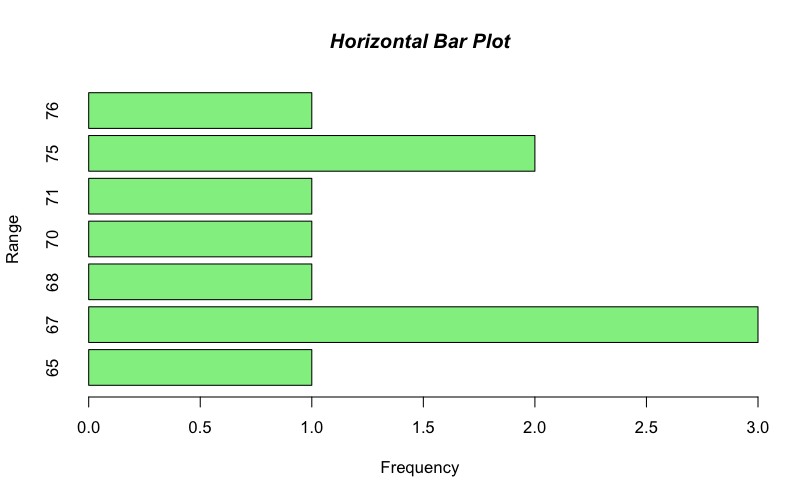
It is straightforward to rotate your plot so that the bars run horizontal rather than vertical (which is the default). To produce a horizontal plot you add horiz= TRUE to the command e.g.
This time I used the title() command to add the main title separately. The value of 4 sets the font to bold italic (try other values).
Histograms
The barplot() function can be used to create a frequency plot of sorts but it does not produce a continuous distribution along the x-axis. A true frequency distribution should have the bar categories (i.e. the x-axis) as continuous items. The frequency plot produced previously had discontinuous categories.
To create a frequency distribution chart you need a histogram, which has a continuous range along the x-axis. The command in R is hist(), and it has various options:
hist(x, breaks, freq, col, ...)
Where:
x – the data to describe, this is usually a single numerical sample (i.e. a vector).
breaks – how to split the break-points. You can give the explicit values (on the x-axis) where the breaks will be, the number of break-points you want, or a character describing an algorithm: the options are “Sturges” (the default), “Scott”, or “FD” (or type “Freedman-Diaconis”).
freq – if set to TRUE the bars show the frequencies. If set to FALSE the bars show density (in which case the total area under the bars sums to 1).
To plot the probabilities (i.e. proportions) rather than the actual frequency you need to add the parameter, freq = FALSE like so:
hist(test.data, freq = FALSE)
You can also use probability = TRUE (instead of freq = FALSE) in the command.
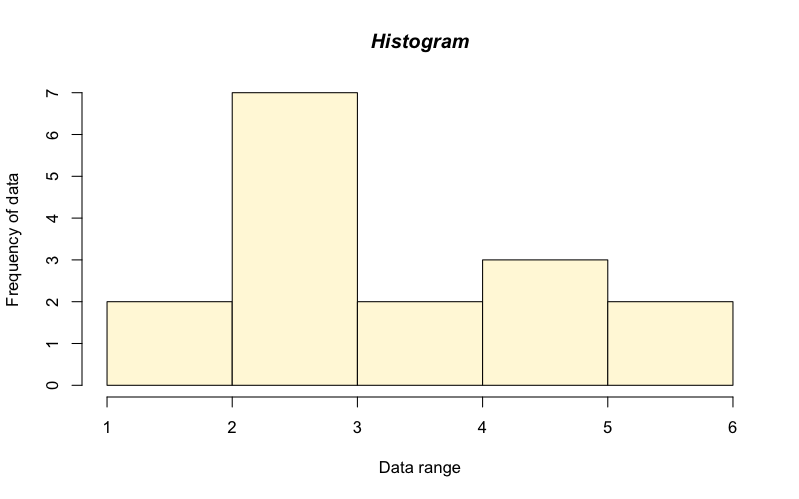
This is useful but the plots are a bit basic and boring. You can change axis labels and the main title using the same commands as for the barplot() command. Here is a new plot with a few enhancements:
> hist(test.data, col = "cornsilk", xlab = "Data range", ylab = "Frequency of data", main = "Histogram", font.main = 4)
These commands are largely self-explanatory. The 4 in the font.main parameter sets the font to italic (try some other values).
By default R works out where to insert the breaks between the bars using the “Sturges” algorithm. You can try other methods:
Using explicit break-points can lead to some “odd” looking histograms, try the examples for yourself (you can copy the data and paste into R)!
Notice how the exact break points are specified in the c(x1, x2, x3) format. You can manipulate the axes by changing the limits e.g. make the x-axis start at zero and run to 6 by another simple command e.g.:
> hist(test.data, 10, xlim=c(0,6), ylim=c(0,10))
This sets 10 break-points and sets the y-axis from 0-10 and the x-axis from 0-6. Notice how the commands are in the format c(lower, upper). The xlim and ylim parameters are useful if you wish to prepare several histograms and want them all to have the same scale for comparison.
Stem-Leaf plots
A very basic yet useful plot is a stem and leaf plot. It is a quick way to represent the distribution of a single sample. The basic command is:
stem(x, scale = 1, ...)
Where:
x – the data to be represented.
scale – how to expand the number of bins presented (default, scale = 1).
The stem() command does not actually make a plot (in that is does not create a plot window) but rather represents the data in the main console.
> test.data
[1] 2.1 2.6 2.7 3.2 4.1 4.3 5.2 5.1 4.8 1.8 1.4 2.5 2.7 3.1 2.6 2.8
> stem(test.data)
The decimal point is at the |
1 | 48
2 | 1566778
3 | 12
4 | 138
5 | 12
The stem-leaf plot is a way of showing the rough frequency distribution of the data. In most cases a histogram would be a better option.
The scale parameter alters the number of rows; it can be helpful to set scale to a larger value than 1 in some cases.
Box-Whisker plots
A box and whisker graph allows you to convey a lot of information in one simple plot. Each sample produces a box-whisker combination where the box shows the extent of the inter-quartiles (that is the 1st and 3rd quartiles), and the whiskers show the max and min values. A stripe is added to the box to show the median. The basic command is boxplot() and it has a range of options:
boxplot(x, range, names, col, horizontal, ...)
Where:
x – the data to plot. This can be a single vector or several (separated by commas). Alternatively you can give a formula of the form y ~ x where y is a response variable and x is a predictor (grouping) variable.
range – the extent of the whiskers. By default values > 1.5 times the IQR from the median are shown as outliers (points). Set range = 0 to get whiskers to go to the full max-min.
names – the names to be added as labels for the boxes on the x-axis.
col – the colour(s) for the boxes.
horizontal – if TRUE the bars are drawn horizontally (but the bottom axis is still considered as the x-axis). The default is FALSE.
… – there are various other options.
The boxplot() command is very powerful and R is geared-up to present data in this form!
Single sample boxplot
You can create a plot of a single sample. If the data are part of a larger dataset then you need to specify which variable to draw:
What you see is a box with a line through it. The line represents the median of the sample. The box itself shows the upper and lower quartiles. The whiskers show the range (i.e. the largest and smallest values). In some cases you may see points in addition to the whiskers:
> boxplot(trees$Volume, col = "lightgreen", ylab = "Value axis", xlab = "Single sample", main = "Box plot with outlier")
Now you see an outlier outside the range of the whiskers. R doesn’t automatically show the full range of data (as I implied earlier). You can control the range shown using a simple parameter range= n. If you set n to 0 then the full range is shown. Otherwise the whiskers extend to n times the inter-quartile range. The default is set to n = 1.5.
> boxplot(trees$Volume, col = "lightgreen", range = 0, ylab = "Value axis", xlab = "Single sample", main = "Box plot with full range")
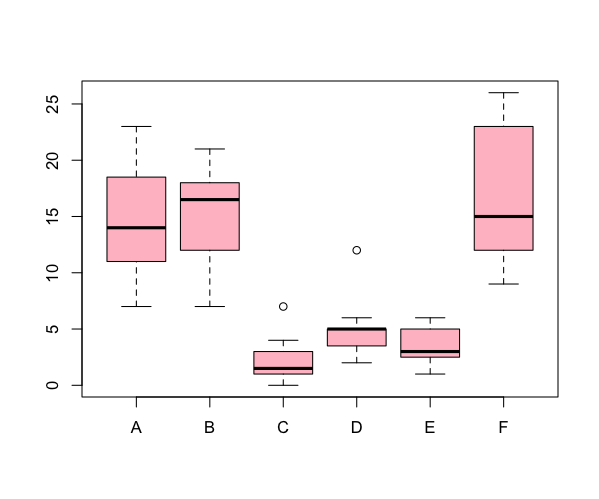
Plotting several samples
If your data contain multiple samples you can plot them in the same chart. The form of the command depends on the form of the data.
In this example the data were arranged in sample layout, so the command only needed to specify the “container”. If the data are set out with separate variables for response and predictor you need a different approach.
> head(InsectSprays)
count spray
1 10 A
2 7 A
3 20 A
4 14 A
5 14 A
These data have a response variable (dependent variable), and a predictor variable (independent variable). You need to specify the data to plot in the form of a formula like so:
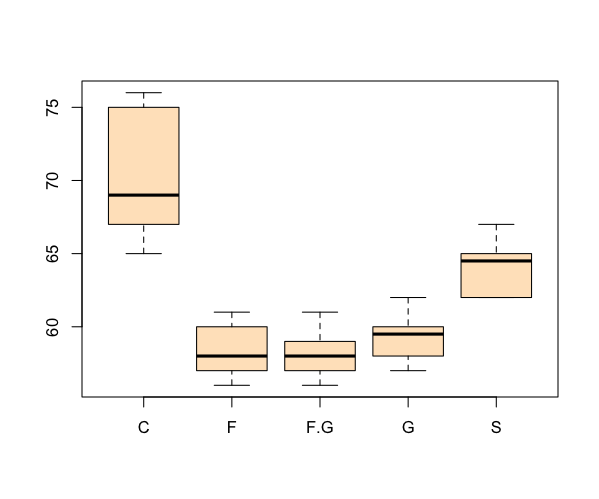
> boxplot(count ~ spray, data = InsectSprays, col = "pink")
The formula is in the form y ~ x, where y is your response variable and x is the predictor. You can specify multiple predictor variables in the formula, just separate then with + signs.
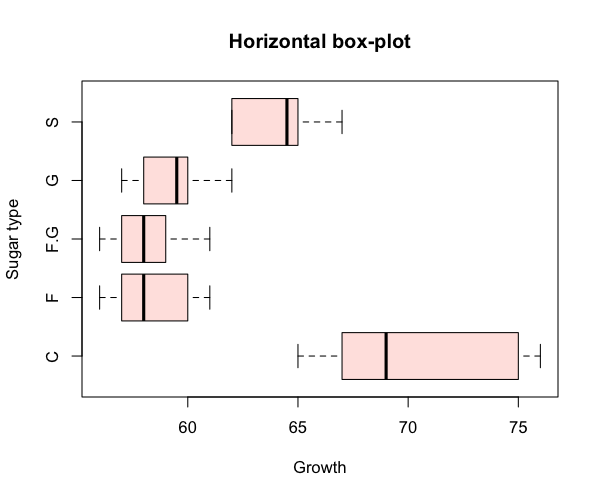
Horizontal box plots
It is straightforward to rotate your plot so that the bars run horizontal rather than vertical (which is the default). To produce a horizontal plot you add horizontal= TRUE to the command e.g.
> boxplot(flies, col = "mistyrose", horizontal = TRUE)
> title(xlab = "Growth", ylab = "Sugar type", main = "Horizontal box-plot")
When you add the titles, either as part of the plotting command or separately via the title() function, you need to remember that ylab is always the vertical (left) axis and xlab refers to the bottom (horizontal) axis.
Scatter plots
A scatter plot is used when you have two variables to plot against one another. R has a basic command to perform this task. The command is plot(). As usual with R there are many additional parameters that you can add to customize your plots.
The basic command is:
plot(x, y, pch, xlab, xlim, col, bg, ...)
Where:
x, y – the names of the variables (you can also use a formula of the form y ~ x to “tell” R how to present the data.
pch – a number giving the plotting symbol to use. The default (1) produces an open circle (try values 0–25).
xlab, ylab – character strings to use as axis labels.
xlim, ylim – the limits of the axes in the form c(start, end).
col – the colour for the plotting symbols.
bg – if using open symbols you use bg to specify the fill (background) colour.
… – there are many additional parameters that you might use.
Here is an example using one of the many datasets built into R:
The default is to use open plotting symbols. You can alter this via the pch parameter. The names on the axes are taken from the columns of the data. If you type the variables as x and y the axis labels reflect what you typed in:
plot(cars$speed, cars$dist)
This command would produce the same pattern of points but the axis labels would be cars$speed and cars$dist. You can use other text as labels, but you need to specify xlab and ylab from the plot() command.
As usual with R there are a wealth of additional commands at your disposal to beef up the display. A useful additional command is to add a line of best-fit. This is a command that adds to the current plot (like the title() command). For the above example you would type:
abline(lm(dist ~ speed, data= cars))
The basic command uses abline(a, b), where a= slope and b= intercept. Here a linear model command was used to calculate the best-fit equation (try typing the lm() command separately, you get the intercept and slope).
If you combine this with a couple of extra lines you can produce a customized plot:
You can alter the plotting symbol using the command pch= n, where n is a simple number. You can also alter the range of the x and y axes using xlim= c(lower, upper) and ylim= c(lower, upper). The size of the plotted points is manipulated using the cex= n parameter, where n = the ‘magnification’ factor. Here are some commands that illustrate these parameters:
Here the plotting symbol is set to 19 (a solid circle) and expanded by a factor of 2. Both x and y axes have been rescaled. The labels on the axes have been omitted and default to the name of the variable (which is taken from the data set).
If you produce a plot you generally get a series of points. The default symbol for the points is an open circle but you can alter it using the pch= n parameter (where n is a value 0–25). Actually the points are only one sort of plot type that you can achieve in R (the default). You can use the parameter type = “type” to create other plots.
“p” – produces points (the default).
“l” – lines only (straight lines connecting the data in the order they are in the dataset).
“b” – points joined with segments of line between (i.e. there are gaps).
“o” – overplot; that is lines with points overlaid (i.e. the line has no gaps).
“n” – nothing is plotted!
So, if your data are “time sensitive” you can choose to display connecting lines and produce some kind of line plot.
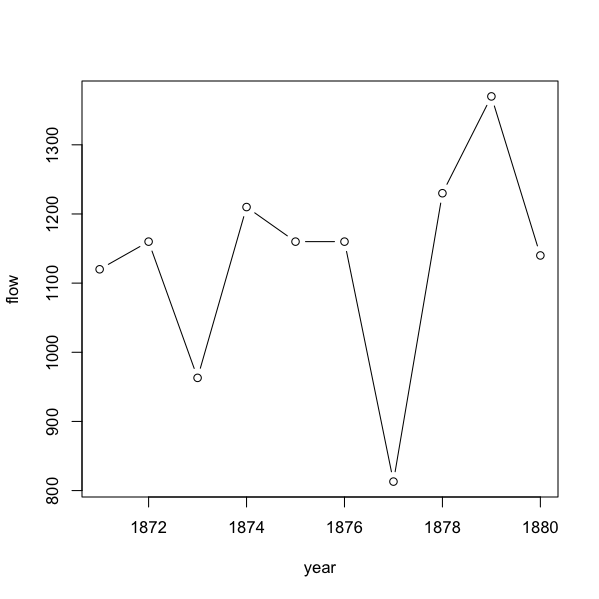
Line plots
You generally use a line plot when you want to “follow” a data series from one interval to another. If your x-data are numeric you can achieve this easily:
Here we use type = “b” and get points with segments of line between them. Note that the x-axis tick-marks line up with the data points. This is unlike an Excel line plot, where the points lie between tick-marks.
In R a line plot is more akin to a scatter plot. In Excel a line plot is more akin to a bar chart.
Custom Axes
If your x-axis data are numeric your line plots will look “normal”. However, if your data are characters (e.g. month names) then you get something different.
> vostok
month temp
1 Jan -32.0
2 Feb -47.3
3 Mar -57.2
4 Apr -62.9
5 May -61.0
6 Jun -70.6
7 Jul -65.5
8 Aug -68.2
9 Sep -63.2
10 Oct -58.0
11 Nov -42.0
12 Dec -30.4
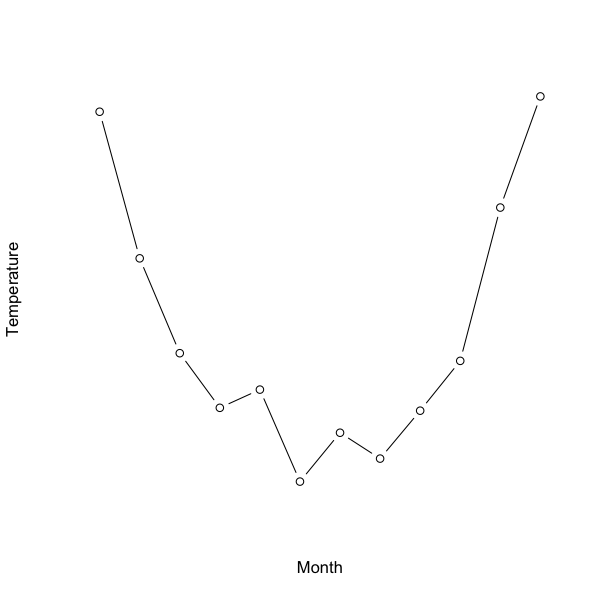
These data show mean temperatures for a research station in the Antarctic. If you attempt to plot the whole variable e.g. plot(temp ~ month) you get a horrid mess (try it and see). This is because the month is a factor and cannot be represented on an x, y scatter plot.
However, if you plot the temperature alone you get the beginnings of something sensible:
> plot(vostok$temp)
So far so good. There appear to be a series of points and they are in the correct order. You can easily join the dots to make a line plot by adding (type= “b”) to the plot command. Notice that the axis label for the x-axis is “Index”, this is because you have no reference (you only plotted a single variable).
What you need to do next is to alter the x-axis to reflect your month variable. You’ll need to make a custom axis with the axis() command but first you need to re-draw the plot without any axes:
The bottom (x-axis) is the one that needs some work. There are 12 values so the at = parameter needs to reflect that. The labels are the month names, which are held in the month variable of the data.
So, the bottom axis ends up with 12 tick-marks and labels taken from the month variable in the original data. Note that here I had to tweak the size of the axis labels with the cex.axis parameter, which made the text a fraction smaller and fitted in the display.
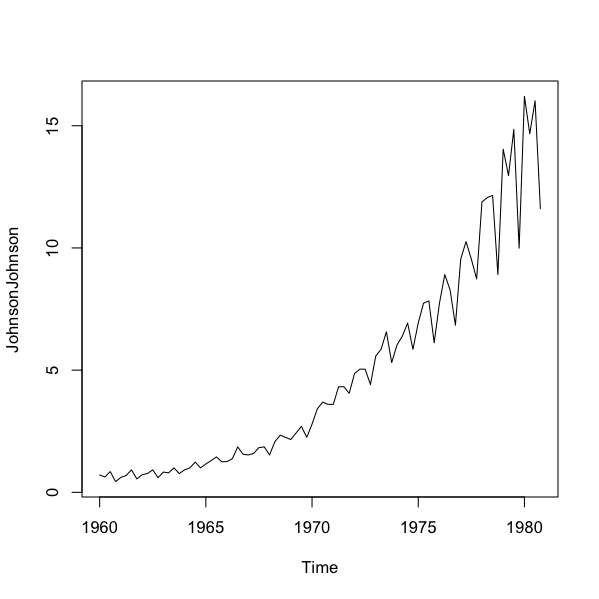
Some datasets are already in a special format called a time-series. R “knows” how the data are split time-wise. Here is an example that is built-in to R”
Time series objects have their own plotting routine and automatically plot as a line, with the labels of the x-axis reflecting the time intervals built into the data:
> plot(JohnsonJohnson)
A time-series plot is essentially plot(x, type = “l”) where R recognizes the x-axis and produces appropriate labels.
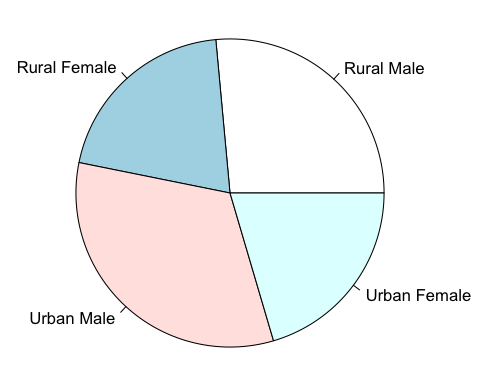
Pie Charts
Pie charts are not necessarily the most useful way of displaying data but they remain popular. You can produce pie charts easily in R using the basic command pie():
pie(x, labels, clockwise, init.angle, col, ...)
Where:
x – the data to plot. This is a single sample (vector) of numbers.
labels – a character string to use for labels (the default takes the names from the data if there are any).
clockwise – the default is FALSE, producing slices of pie in a counterclockwise (anticlockwise) direction.
angle – the starting point for the first slice of pie. The default is 90 (degrees) if plotting anticlockwise and 0 if clockwise.
col – colours to use for the pie slices. If you specify too few colours they are recycled and if you specify too many some are not used. The default colours are pastel shades.
… – there are several additional parameters you could use.
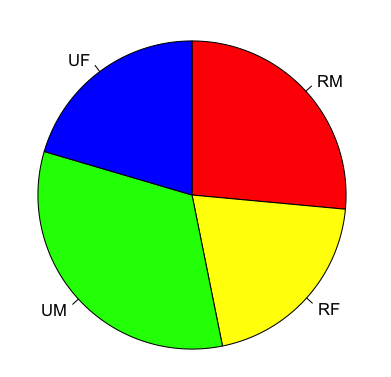
A basic pie chart could be made so:
> VADmeans = colMeans(VADeaths)
> VADmeans
Rural Male Rural Female Urban Male Urban Female
32.74 25.18 40.48 25.28
> pie(VADmeans)
You can alter the labels used and the colours as well as the direction the pie is drawn:
Setting the starting angle is slightly confusing (well, I am always confused). The init.angle parameter requires a value in degrees and 90 degrees is 12 o’clock (0 degrees is 3 0’clock).
You could think of regression as like an extension of correlation but where you have a definite response (dependent) variable and at least one predictor (independent) variable. Where you have multiple predictor variables you use multiple regression. In basic linear regression you are using the properties of the normal distribution to tell something about the relationship between the response and the various predictors.
The process is often called regression modelling or linear modelling and is carried out in R with the lm() command.
Linear Regression Models
The lm() function requires a formula that describes the experimental setup. It is generally a good idea to assign a named object to hold the result, as it contains useful components:
Residual standard error: 23.5 on 108 degrees of freedom
(42 observations deleted due to missingness)
Multiple R-squared: 0.5103, Adjusted R-squared: 0.5012
F-statistic: 56.28 on 2 and 108 DF, p-value: < 2.2e-16
The summary() command gives a standard sort of report. The section labelled Coefficients: gives the basic regression information about the various terms of the regression model. There is a row for each predictor plus one for the intercept.
The bottom part of the summary gives the overall model “result”.
Regression coefficients
Once you have a regression model result you can “extract” the coefficients (that is the slopes and intercept) with the coef() command:
The regular coefficients are in units related to the original variables.
Beta coefficients
R does not compute beta coefficients as standard. In a regression the beta coefficients are standardized against one another and are therefore in units of standard deviation. This allows you to compare variables.
You can calculate a beta coefficient like so:
beta = coeff * SD(x) / SD(y)
Where SD is standard deviation.
In this dataset there are some missing values so to get the standard deviation:
Correlation is when you are looking to determine the strength of the relationship between two numerical variables. R can carry out correlation via the cor() command, and there are three different sorts:
Pearson correlation – for where data are normally distributed.
Spearman’s Rank (or Rho) – for where data are non-parametric (not normally distributed).
Kendall’s Tau – for where data are non-parametric.
To carry pout the correlation you need two variables to compare:
cor(x, y, method = "pearson")
The default is to use Pearson’s product moment but you can specify “spearman” or “kendall” to carry out the appropriate calculation.
The matrix shows the correlation of every variable with every other variable.
Correlation and Significance tests
The basic cor() function computes the strength (and direction) of the correlation but does not tell you if the relationship is statistically significant. You need the cor.test() command to carry out a statistical test.
> cor.test(fw$count, fw$speed)
Pearson’s product-moment correlation
data: fw$count and fw$speed
t = 2.5689, df = 6, p-value = 0.0424
alternative hypothesis: true correlation is not equal to 0
95 percent confidence interval:
0.03887166 0.94596455
sample estimates:
cor
0.7237206
You can also specify the variables in a formula like so:
> cor.test(~ Length + Algae, data = mf, method = "spearman")
Spearman’s rank correlation rho
data: Length and Algae
S = 517.65, p-value = 1.517e-06
alternative hypothesis: true rho is not equal to 0
sample estimates:
rho
0.8009031
Notice that the formula starts with a tilde ~ and then you provide the two variables, separated with a +. This arrangement reinforces the notion that you are looking for a simple correlation and are not implying cause and effect (there is no response ~ predictor in the formula).
When you have more than two samples to compare your go-to method of analysis would generally be analysis of variance (see 15). However, if your data are not normally distributed you need a non-parametric method of analysis. The Kruskal-Wallis test is the test to use in lieu of one-way anova.
Kruskal Wallis test
The kruskal.test() command will carry out the Kruskal Wallis test for you.
In this case you see that the data contains five columns, each being a separate sample. If your data are in separate named objects you can still run the test but you bundle the data in a list():
> Grass ; Heath ; Arable
[1] 3 4 3 5 6 12 21 4 5 4 7 8
[1] 6 7 8 8 9 11 12 11 NA NA NA NA
[1] 19 3 8 8 9 11 12 11 9 NA NA NA
> kruskal.test(list(Grass, Heath, Arable))
Kruskal-Wallis rank sum test
data: list(Grass, Heath, Arable)
Kruskal-Wallis chi-squared = 6.0824, df = 2, p-value = 0.04778
Kruskal Wallis and stacked data
The kruskal.test() function is also able to deal with data that are in a different form.
> fly
size diet
1 75 C
2 67 C
3 70 C
4 75 C
5 65 C
6 71 C
In this case one column gives the response variable and the other gives the (grouping) predictor variable.
> attach(fly)
> kruskal.test(size, g = diet)
Kruskal-Wallis rank sum test
data: size and diet
Kruskal-Wallis chi-squared = 38.437, df = 4, p-value = 9.105e-08
> detach(fly)
Note that attach() was used here to allow R to “read” the two columns from the data. More conveniently you can use a formula to describe the situation. This means you do not need to use attach().
> bfs
count site
1 3 Grass
2 4 Grass
3 3 Grass
4 5 Grass
5 6 Grass
6 12 Grass
> kruskal.test(count ~ site, data = bfs)
Kruskal-Wallis rank sum test
data: count by site
Kruskal-Wallis chi-squared = 6.0824, df = 2, p-value = 0.04778
A formula is a powerful tool in R and is used in many functions. You put the name of the response variable first then a ~ followed by the predictor, finally you add the data part that R knows where to find the variables.
Post Hoc test for Kruskal Wallis
The Kruskal Wallis test will give you an overall result (both the examples show significant differences between groups). However, you will want to know about the details of the differences between groups. To do this you carry out a post hoc test. This means that you take the groups and compare them pair by pair. In this way you explore the data more fully and can describe exactly how each group relates to the others.
You cannot simply conduct several two-sample tests, multiple U-tests for example, as you increase your chances of getting a significant result by pure chance. For parametric analyses there are “standard” methods for taking this multiple testing into account. However, for Kruskal Wallis there is no consensus about the “right” method!
The simplest method is to carry out regular U-tests but correct for the use of multiple analyses. Such corrections are often called “Bonferroni” corrections, although there are other methods of correction. The pairwise.wilcox.test() function can conduct pairwise U-tests and correct the p-values for you.
You need to specify the data and the grouping:
> head(fly, n = 4)
size diet
1 75 C
2 67 C
3 70 C
4 75 C
> pairwise.wilcox.test(fly$size, g = fly$diet)
Pairwise comparisons using Wilcoxon rank sum test
data: fly$size and fly$diet
C F F.G G
F 0.0017 – – –
F.G 0.0017 1.0000 – –
G 0.0017 0.4121 0.2507 –
S 0.0027 0.0017 0.0017 0.0017
P value adjustment method: holm
The command carries out multiple U-tests and adjusts the p-values. In this case the “holm” method of adjustment was used (the default). You can select others:
> head(bfs)
count site
1 3 Grass
2 4 Grass
3 3 Grass
4 5 Grass
5 6 Grass
6 12 Grass
> pairwise.wilcox.test(bfs$count, g = bfs$site, p.adjust.method = "bonferroni")
Pairwise comparisons using Wilcoxon rank sum test
data: bfs$count and bfs$site
Arable Grass
Grass 0.16 –
Heath 1.00 0.11
P value adjustment method: bonferroni
Try help(p.adjust.methods) from R for more information about the different adjustment methods.
Friedman test
The Friedman test is essentially a 2-way analysis of variance used on non-parametric data. The test only works when you have completely balanced design. You can also use Friedman for one-way repeated measures types of analysis.
Here is an example of a data file which has a 2-way unreplicated block design.
What you have here are data on surveys of amphibians. The first column (count) represents the number of individuals captured. The final column is the year that the survey was conducted. The middle column (month) shows that for each year there were 5 survey events in each year. What you have here is a replicated block design. Each year is a “treatment” (or “group”) whilst the month variable represents a “block”. This is a common sort of experimental design; the blocks are set up to take care of any possible variation and to provide replication for the treatment. In this instance you wish to know if there is any significant difference due to year.
The Friedman test allows you to carry out a test on these data. You need to determine which variable is the group and which the block. The friedman.test() function allows you to perform the test. One way is to specify the groups and the blocks like so:
> attach(survey)
> friedman.test(count, groups = year, blocks = month)
Friedman rank sum test
data: count, year and month
Friedman chi-squared = 7.6, df = 2, p-value = 0.02237
> detach(survey)
The other method is using a formula syntax:
> friedman.test(count ~ year | month, data= survey)
Here the vertical bar “splits” the predictor variables into group | block. It is important to get the group and block variables the correct way around!
Post Hoc testing for Friedman tests
There is a real drop in statistical power when using a Friedman test compared to anova. Although there are methods that enable post-hoc tests, the power is such that obtaining significance is well-nigh impossible in most cases.
You can try pairwise.wilcox.test() with various groupings, e.g.:
> pairwise.wilcox.test(survey$count, g = survey$year)
Pairwise comparisons using Wilcoxon rank sum test
data: survey$count and survey$year
2004 2005
2005 0.033 –
2006 0.033 0.834
P value adjustment method: holm
In this analysis any other grouping is impossible (as there is no replication).
The analysis of variance is a commonly used method to determine differences between several samples. R provides a method to conduct ANOVA via the aov() function. Your data need to be arranged in a particular manner for the aov() command to “work” (see 13). Your data need to be set out so that each column represents a single variable like so:
> head(fly)
size diet
1 75 C
2 67 C
3 70 C
4 75 C
5 65 C
6 71 C
In this example the first column (size) is the response variable (dependent variable) and the second column (diet) is the predictor (independent) variable. ANOVA is a method of comparing samples that are grouped. The simplest arrangement is where you have a single grouping variable (predictor); this is called one-way ANOVA. If you have two predictor variables the analysis is called two-way ANOVA and so on.
The aov() command requires a formula that describes the experimental setup. In general terms you have:
response ~ predictor
This describes a 1-way anova.
ANOVA One-way
In one-way anova you have a response (dependent) variable and one predictor (grouping or independent) variable. It is usually best to assign a named object to “hold” the result of aov(), as there are multiple components you can make use of.
> mod = aov(size ~ diet, data = fly)
> summary(mod)
Df Sum Sq Mean Sq F value Pr(>F)
diet 4 1077.3 269.33 49.37 6.74e-16 ***
Residuals 45 245.5 5.46
—
Signif. codes: 0 ‘***’ 0.001 ‘**’ 0.01 ‘*’ 0.05 ‘.’ 0.1 ‘ ’ 1
The summary() command produces an anova table, a standard way to present such results. The table gives all the information you require.
Post hoc testing
The basic ANOVA produces an overall result. In the previous example you can see a significant result; the different diets produce different sizes of fly wings! However, this is not the complete story, are all the groups different from one another or are some different and some not? What you need is a post hoc test. This carries out pair by pair analyses and takes into account the fact that you are running multiple tests (and so more likely to get a significant result by chance).
There are a number of different methods but the Tukey HSD test is the most common, and the TukeyHSD() command is built-in to R. You simply run the command on the result of aov(). In this case I used ordered = TRUE to make the results more readable:
The ordered = TRUE parameter makes sure that the pairs are compared in an order that ensures that the differences in means are always positive. So for example, F-C gives 11.9 as a mean difference but C-F would be -11.9. This simply makes the result easier to read.
the result shows the difference in mean value as well as 95% confidence intervals. The final column gives an adjusted p-value (the significance). You can see that some of the comparisons are not significant (values > 0.05), whilst others are (values < 0.05).
ANOVA models
The aov() command uses a formula to describe the setup. The formula is a powerful syntax that allows you to specify complicated anova models. Here are just a few examples:
Model
Meaning
y ~ a
A one-way anova where a is a predictor (grouping variable).
y ~ a + b
A two-way anova where a and b are different predictors acting independently.
y ~ a * b
A two-way anova with a and b as predictors but where there is also an interaction between them.
y ~ a + b + a:b
The same as above but with the interaction written explicitly.
y ~ a + b + Error(c)
A two-way anova with a and b as predictors. Another variable, c, forms the error term, that is the within group variation. This is the way to describe repeated measures anova models.
y ~ a * b + Error(c/b)
This is a two-way repeated measures anova where b is nested in c and the predictors a and b have interaction.
So, the formula syntax can describe quite complicated situations! The preceding table shows but a few of the options.
The functions operate over a one-dimensional object (called a vector in R-speak). If your data are a column of a larger dataset then you’ll need to use attach() or the $ so that R can “find” your data.
If your data contain NA elements, the result may show as NA. You can overcome this in many (but not all) commands by adding na.rm = TRUE as a parameter.
> nad
[1] NA 3 5 7 5 3 2 6 8 5 6 9 NA NA
> mean(nad)
[1] NA
> mean(nad, na.rm = TRUE)
[1] 5.363636
T-test
Student’s t-test is a classic method for comparing mean values of two samples that are normally distributed (i.e. they have a Gaussian distribution). Such samples are described as being parametric and the t-test is a parametric test. In R the t.test() command will carry out several versions of the t-test.
t.test(x, y, alternative, mu, paired, var.equal, …)
x – a numeric sample.
y – a second numeric sample (if this is missing the command carries out a 1-sample test).
alternative – how to compare means, the default is “two.sided”. You can also specify “less” or “greater”.
mu – the true value of the mean (or mean difference). The default is 0.
paired – the default is paired = FALSE. This assumes independent samples. The alternative paired = TRUE is used for matched pair tests.
equal – the default is var.equal = FALSE. This treats the variance of the two samples separately. If you set var.equal = TRUE you conduct a classic t-test using pooled variance.
… – there are additional parameters that we aren’t concerned with here.
In most cases you want to compare two independent samples:
> mow
[1] 12 15 17 11 15
> unmow
[1] 8 9 7 9
> t.test(mow, unmow)
Welch Two Sample t-test
data: mow and unmow
t = 4.8098, df = 5.4106, p-value = 0.003927
alternative hypothesis: true difference in means is not equal to 0
95 percent confidence interval:
2.745758 8.754242
sample estimates:
mean of x mean of y
14.00 8.25
If you specify a single variable you can carry out a 1-sample test by specifying the mean to compare against:
> data1
[1] 3 5 7 5 3 2 6 8 5 6 9
> t.test(data1, mu = 5)
One Sample t-test
data: data1
t = 0.55902, df = 10, p-value = 0.5884
alternative hypothesis: true mean is not equal to 5
95 percent confidence interval:
3.914249 6.813024
sample estimates:
mean of x
5.363636
If you have matched pair data you can specify paired = TRUE as a parameter (see more in 14.4).
U-test
The U-test is used for comparing the median values of two samples. You use it when the data are not normally distributed, so it is described as a non-parametric test. The U-test is often called the Mann-Whitney U-test but is generally attributed to Wilcoxon (Wilcoxon Rank Sum test), hence in R the command is wilcox.test().
wilcox.test(x, y, alternative, mu, paired, …)
x – a numeric sample.
y – a second numeric sample (if this is missing the command carries out a 1-sample test).
alternative – how to compare means, the default is “two.sided”. You can also specify “less” or “greater”.
mu – the true value of the median (or median difference). The default is 0.
paired – the default is paired = FALSE. This assumes independent samples. The alternative paired = TRUE is used for matched pair tests.
… – there are additional parameters that we aren’t concerned with here.
> wilcox.test(Grass, Heath)
Wilcoxon rank sum test with continuity correction
data: Grass and Heath
W = 20.5, p-value = 0.03625
alternative hypothesis: true location shift is not equal to 0
If there are tied ranks you may get a warning message about exact p-values. You can safely ignore this!
Paired tests
The t-test and the U-test can both be used when your data are in matched pairs. Sometimes this kind of test is also called a repeated measures test (depending on circumstance). You can run the test by adding paired = TRUE to the appropriate command.
Here is an example where the data show the effectiveness of greenhouse sticky traps in catching whitefly. Each trap has a white side and a yellow side. To compare white and yellow we can use a matched pair.
> mpd
white yellow
1 4 4
2 3 7
3 4 2
4 1 2
5 6 7
6 4 10
7 6 5
8 4 8
> attach(mpd)
> t.test(white, yellow, paired = TRUE, var.equal = TRUE)
Paired t-test
data: white and yellow
t = -1.6567, df = 7, p-value = 0.1415
alternative hypothesis: true difference in means is not equal to 0
95 percent confidence interval:
-3.9443258 0.6943258
sample estimates:
mean of the differences
-1.625
> detach(mpd)
You can do a similar thing with the wilcox.test().
Chi Squared tests
Tests for association are easily carried out using the chisq.test() command. Your data need to be arranged as a contingency table. Here is an example:
In this dataset the columns form one set of categories (habitats) and the rows form another set (bird species). In the original spreadsheet (CSV file) the first column contains the bird species names; these are “converted” to row names when the data are imported:
You can carry out a test for association with the chisq.test() function. This time though, you should assign the result to a named variable.
> cs = chisq.test(bird)
Warning message:
In chisq.test(bird) : Chi-squared approximation may be incorrect
> cs
Pearson’s Chi-squared test
data: bird
X-squared = 78.274, df = 20, p-value = 7.694e-09
In this instance you get a warning message (this is because there are expected values < 5). The basic “result” shows the overall significance but there are other components that may prove useful:
Now you can see the expected values. Other useful components are $residuals and $stdres, which are the Pearson residuals and the standardized residuals respectively.
You can also use square brackets to get part of the result e.g.
Now you can see the standardized residuals for the Blackbird row.
Yates’ correction
When using a 2 x 2 contingency table it is common practice to reduce the Observed – Expected differences by 0.5. To do this add correct=TRUE to the original function e.g.
> your.chi = chisq.test(your.data, correct=TRUE)
If your table is larger than 2 x 2 then the correction will not be applied (the basic test will run instead) even if you request it.
Goodness of Fit test
A goodness of fit test is a special kind of test of association. You use it when you have a set of data in categories that you want to compare to a “known” standard. A classic example is in genetics, where the theory suggests a particular ratio of phenotypes:
> pea
[1] 116 40 31 13
> ratio
[1] 9 3 3 1
Here you have counts of pea plants (there are 4 phenotypes) from an experiment in cross-pollination. The genetic theory suggests that your results should be in the ratio 9:3:3:1. Are these results in that ratio? The goodness of fit test will tell you.
> gfit = chisq.test(pea, p = ratio, rescale.p = TRUE)
> gfit
Chi-squared test for given probabilities
data: pea
X-squared = 1.4222, df = 3, p-value = 0.7003
Notice that the rescale.p = TRUE parameter was used to ensure that the expected probabilities sum to one. The final result is not significant, meaning that the peas observed are not statistically significantly different from the expected ratio.
The result contains the same components as the regular chi squared test:
The items you want to join are listed in the parentheses and separated with commas. The items are joined in the order they appear in the command.
Typing in values using scan()
The c() command is useful but it can be a pain to type all the commas! The scan() command allows you to enter data from the keyboard without commas. Here are some of the possible parameters:
scan(what, sep, dec = “.”)
what – the default is to expect numbers but you can also choose what = “character” for text.
sep – the default is to use a space to separate elements, use sep = “,” for commas or sep = “\t” for tabs.
dec – the decimal point character defaults to a full stop (period).
To use scan() you start by assigning a name to “hold” the result. At the same time you tell the command the type of data to expect and if the elements are separated by anything other than spaces.
> mydata = scan()
1:
R now “waits” and you will see the 1: instead of the regular cursor. You can now type your data (in this example it will be expecting numbers).
1: 2 5 6.2 33 25 1.3 8
If you press <enter> then R continues on a new line but still waits:
8:
Type in more data if you like:
8: 111
To stop adding values you press <enter> on a blank line (in other words press <enter> twice).
9:
Read 8 items
You can now see your data by typing the name you assigned to it:
> mydata
[1] 2.0 5.0 6.2 33.0 25.0 1.3 8.0 111.0
If you want to type text values, then use what = “character” as a parameter:
> myscan = scan(what = "character")
1: jan feb mar apr
5: may jun
7:
Read 6 items
> myscan
[1] "jan" "feb" "mar" "apr" "may" "jun"
Note that you don’t have to use quotes around the items as you type (this is way you used what = “character”).
Read the clipboard
The scan() command can read the clipboard, which means you can copy and paste items from other programs (or web pages). Use the sep parameter to match the separator.
Importing data
The next step is to get your data into R. If you have saved your data in a .CSV file then you can use the read.csv(filename) command to get the information. You need to tell R where to store the data and to do this you assign it a name. All names must have at least one letter (otherwize it is a number of course!). Remember, you can use a period (e.g. test.1) or underscore (e.g. test_1) but no other punctuation marks. R is case sensitive so the variable test is different from Test or teSt.
The read.csv() command has various parameters that allow you control over how and what is imported, here are some of the essentials:
file – the filename of the data you want to read in. Give this in quotes, with the full path e.g. “My documents/mydata.csv”. Alternatively you can use choose() to bring up a file explorer, allowing you to choose the file.
header – if set to TRUE (the default) the first row of the CSV is used as column labels. You can set this to FALSE, then R will assume all the data are data and assign standard names for the columns (e.g. V1, V2, V3).
sep – this sets the separator between “fields”, given as a character in quotes. For a CSV this is of course a comma (this is the default). If you have tab separated data use sep = “\t”
dec – this sets the decimal point character. The default is a full stop (period) dec = “.” but other characters are possible.
names – this allows a column of data to be used as labels (i.e. row names). Usually this would be the first column e.g. row.names = 1, but any number is allowed.
Here are some examples:
To get a file into R with basic columns of data and their labels use:
> mydata = read.csv(file.choose(), header = TRUE)
To get a file into R with column headings and row headings use:
> mydata = read.csv(file.choose(), row.names = 1)
To read a CSV file from a European colleague where commas were used as decimal points and the data were separated by semi-colon.
> mydata = read.csv(file.choose(), dec = ",", sep = ";")
On Linux machines the file.choose() capability is absent, so you’ll need to use the full file and path name (in quotes):
> mydata = read.csv(file = "My Documents/myfile.CSV")
N.B. There are occasions when R won’t like your data file! Check the file carefully. In some cases, the addition of an extra linefeed at the end will sort out the issue. To do this open the file in a word processor and make sure that non-printing characters are displayed. Add the extra carriage return and save the file.
Seeing your data in R
To view your data once it is imported you simply type the name you assigned to it.
In this case the first column of data was set to act as row names, so when you view the data you see those names. In other cases you do not have explicit row names, in which case R assigns a simple index number just to help you navigate the dataset.
Notice that each column has a name (taken from the CSV) but that simple numbers are used as a “row index”.
View individual data columns
The variable name you assign to data that you import “covers” the overall data. However, R cannot pick out individual columns (variables) in your data without a bit of help.
> Algae
Error: object ‘Algae’ not found
There are several ways to overcome this, here are two of them:
Use the $ and specify the overall data and column.
Use the attach() command to “open up” the data.
The $ is a general part of the R language and allows you to specify a variable inside another named object like so:
> mf$Algae
[1] 40 45 45 80 75 65 65 65
The attach() command allows the names of the columns in your data to be “seen” by R:
> attach(mf)
> Speed
[1] 12 14 12 16 20 21 17 14
But beware! You may have other data with the same names. To avoid “confusion” you should use detach() after you are done:
> detach(mf)
> Algae
Error: object ‘Algae’ not found
View individual data rows
The $ syntax allows you to “pick out” named elements of a dataset (e.g. the columns) but cannot help you extract individual rows. Instead you can use square brackets to extract one or more elements from a larger item.
data[rows, columns]
If your data are two-dimensional (i.e. have rows and columns) then you always specify the rows first and then the columns. If you leave out one of these, you get “everything”.
When you import data from a file (e.g. CSV) R “tidies up” and makes the resulting data a nice rectangular shape. If any data column is shorter than the others it will be padded with NA items.
In this example the mow column has 5 elements but the unmow column contains only 4. R has “interpreted” the empty cell as a missing value and replaced it with NA (think of it as “not available”).
The NA item is important in data analysis. In this case the NA is obviously simply the result of a “short” column. In other datasets NA may well represent missing values (R will replace any missing cells of a spreadsheet with NA). Some statistical tests will return a result of NA if the data contains NA items. In most cases you may ignore the NA values by including the parameter na.rm= TRUE.
Stacking data
Sometimes you need to reorganize your data and convert from one kind of recording layout to another. For example:
This dataset shows five columns. Each column shows repeated measurements of the size of fly wings under a specific diet. If you want to compare the diets, you’ll need to carry out analysis of variance but R will need the data in a different form.
size diet
1 75 C
2 67 C
3 70 C
4 75 C
5 65 C
6 71 C
In this layout the numerical data are all in one column, called size. This is the response variable (sometimes called dependent variable). The second column is headed diet, and is a grouping variable (each size measurement is related to a group), where the names of the groups are related to the column headings in the previous layout. This column is the predictor variable (also called independent variable).
The stack() command allows you to reassemble data in multi-sample layout to a stacked layout.
> fly = stack(flies)
fly
values ind
1 75 C
2 67 C
3 70 C
4 75 C
5 65 C
6 71 C
Notice that the numerical variable is called values and the grouping variable is called ind.
Selecting columns
You do not have to use all the columns when you stack() the data; use the select parameter to choose:
> stack(flies, select = c(“F”, “G”, “S”))
values ind
1 58 F
2 61 F
3 56 F
4 58 F
5 57 F
6 56 F
Naming the stacked columns
You can carry out your analyses using the names R has used when it did the stack(). If you want to use different names then use the names() command:
> names(fly) = c(“size”, “diet”)
size diet
1 75 C
2 67 C
3 70 C
Note that you give the names wrapped in a c() command and the names themselves in quotes.
Unstack
The opposite of the stack() command is unstack(). This looks to reassemble the data so that each grouping level is matched up with the appropriate values. If there are equal replicates in each grouping, then your result is a neat data frame:
> unstack(fly)
C F F.G G S
1 75 58 58 57 62
2 67 61 59 58 66
3 70 56 58 60 65
4 75 58 61 59 63
If the replication is unbalanced the result is several individual elements bundled together in a list.
In this example the grass dataset shows that there are 5 replicates for mow and 4 for unmow.
What data are loaded?
After a while you may well end up with quite a few items in R. It is easy to lose track of what data are loaded (in computer memory) into your R console (the name of the main window where you type stuff). The ls() command shows you what items are currently available:
Note how the “index” on the left helps you to see how many things there are.
You can also see what items are contained in each object by using ls() and specifying an object:
> ls(mf)
[1] "Algae" "BOD" "Length" "NO3" "Speed"
Removing data items
The rm() command allows you to remove items from the computer memory and so also from the R console. Just place the names of the items to “delete” in the parentheses, separated by commas:
rm(name1, name2, name3, …)
On Windows and Mac computers the GUI also has toolbar menus you can click to remove everything. Use this with caution as there is no undo button!
Once you have installed R and run the program you will see an opening window and a message along these lines:
R : Copyright 2006, The R Foundation for Statistical Computing
Version 2.3.1 (2006-06-01)
ISBN 3-900051-07-0
R is free software and comes with ABSOLUTELY NO WARRANTY.
You are welcome to redistribute it under certain conditions.
Type ‘license()’ or ‘licence()’ for distribution details.
Natural language support but running in an English locale
R is a collaborative project with many contributors.
Type ‘contributors()’ for more information and
‘citation()’ on how to cite R or R packages in publications.
Type ‘demo()’ for some demos, ‘help()’ for on-line help, or
‘help.start()’ for an HTML browser interface to help.
Type ‘q()’ to quit R.
[Previously saved workspace restored]
>
The > is the “prompt”, this is the point where you type in commands (or paste them in from somewhere else). The window you see is part of the GUI and some operations are possible from the menus (including quit). You will generally be asked if you wish to save the workspace. R stores a list of commands and any data sets that are loaded. It can be pretty useful to say “yes” and to save the workspace. The command history is available by using the up and down arrows. You can easily scroll back through previous commands and edit them if needed. You can copy items from previous commands or in fact from any window on the screen and paste them into the current command line. You can also use the left and right arrow keys to move through the current command.
Simple maths
You can think of R as like a big calculator. You type in some maths and get the results.
> 1 + 2 * 3
[1] 7
Note that the calculations are carried out in the “standard” manner, with multiplications and divisions before additions or subtractions. Anything in brackets (parentheses) is computed before things outside:
> (1 + 2) * 3
[1] 9
Your result is displayed immediately. Often you will want to store the results to use later, so you need to assign a name to the result.
> result1 = 12 + 17 / 2
> result1
[1] 20.5
Notice that you do not see the result right away, you must type the name to view it. The result can be used for more calculations:
You can use all the “regular” letters of the alphabet a-z (or A-Z) as well as numbers 0-9. R is case sensitive so the following names are different!
result1
Result1
RESULT1
You also use the full stop (period) and underscore characters. You must start a name with a letter and not a number.
Assignment
In the examples here I used = to “define” a result. You will often see something like this <- which looks a bit like an arrow. This is the “preferred” way to assign names to results and you’ll see this in the R help entries and examples.