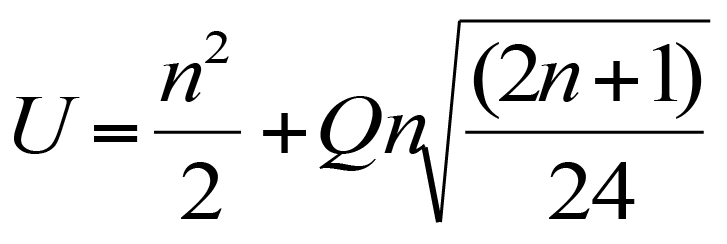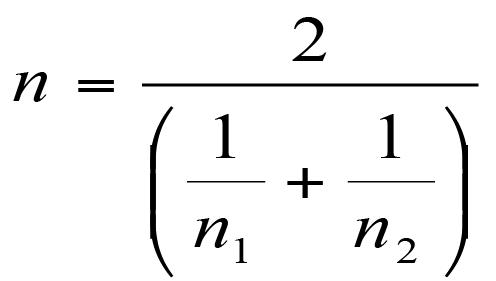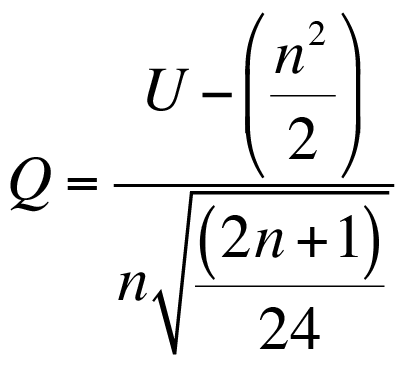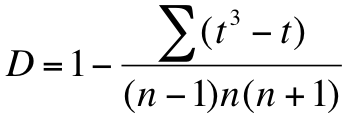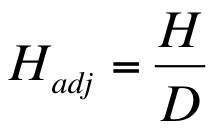Exercise 12.1.4.
Statistics for Ecologists (Edition 2) Exercise 12.1.4
This exercise is concerned with comparing diversity (Section 12.1.4) and in particular how to carry out a modified version of the t-test designed to compare the Shannon diversity index of two samples.
Comparing the Shannon diversity index of two samples using Hutcheson t-test
Introduction
When you have two samples of community data you can calculate a diversity index for each one. The Shannon diversity index is a commonly used measure of diversity. However, you cannot compare the two index values using classic hypothesis tests because you do not have replicated data.
The Hutcheson t-test is a modified version of the classic t-test that provides a way to compare two samples. The key is the formula that determines the variance of the Shannon index. These notes will show you how to conduct the Hutcheson t-test and so get a statistical significance of the difference in Shannon diversity between two samples. There is also a spreadsheet calculator, that you can download and use for your own data.
The Hutcheson t-test
The Hutcheson t-test was developed as a method to compare the diversity of two community samples using the Shannon diversity index (Hutcheson 1970, J. Theor. Biol. 29 p.151). The basic formula is similar in appearance to the classic t-test formula.

Formula for the Hutcheson t-test.
In the formula H represents the Shannon diversity index for each of the two samples (subscripted a and b). The bottom of the formula refers to the variance of each of the samples.
Computing variance for the Shannon diversity index
Computing the variance of the Shannon diversity is done using the formula shown below.

Formula to calculate variance in Shannon diversity. S is the species richness; N is the total number of individuals.
In the formula S is the total number of species, whilst N is the total abundance. The p is the proportion that each species makes towards the total. The formula is fairly easy to evaluate; it looks fairly horrid but the components are easily computed.
Getting a t-value
It is easiest to use a spreadsheet for the calculations. The computations are relatively straightforward. You can use the example spreadsheet Shannon diversity t-test calculator.xlsx rather than make your own! Here is an example of how the calculations appear in the spreadsheet.
Example of calculations in Shannon diversity t-test spreadsheet.
| Site Name: | E1 | ||||
| Taxon | Count | P | Ln(P) | P*Ln(P) | P*Ln(P)2 |
| 388 | 0.543 | -0.611 | -0.332 | 0.203 | |
| 1 | 0.001 | -6.572 | -0.009 | 0.060 | |
| 2 | 0.003 | -5.879 | -0.016 | 0.097 | |
| 13 | 0.018 | -4.007 | -0.073 | 0.292 | |
| 3 | 0.004 | -5.474 | -0.023 | 0.126 | |
| 1 | 0.001 | -6.572 | -0.009 | 0.060 | |
| 59 | 0.083 | -2.495 | -0.206 | 0.514 | |
| 3 | 0.004 | -5.474 | -0.023 | 0.126 | |
| 2 | 0.003 | -5.879 | -0.016 | 0.097 | |
| 2 | 0.003 | -5.879 | -0.016 | 0.097 | |
| 210 | 0.294 | -1.225 | -0.360 | 0.441 | |
| 4 | 0.006 | -5.186 | -0.029 | 0.150 | |
| 3 | 0.004 | -5.474 | -0.023 | 0.126 | |
| 1 | 0.001 | -6.572 | -0.009 | 0.060 | |
| 1 | 0.001 | -6.572 | -0.009 | 0.060 | |
| 21 | 0.029 | -3.528 | -0.104 | 0.366 | |
| 1 | 0.001 | -6.572 | -0.009 | 0.060 | |
| 0 | |||||
| 0 | |||||
| 0 | |||||
| Total | 715 | 1 | -83.972 | -1.267 | 2.934 |
| Richness | 17 | ||||
| SS | 2.934 | ||||
| SQ | 1.606 | ||||
| H | 1.267 | ||||
| S2H | 0.002 |
The species names are not essential but the spreadsheet has room for them. You need the total abundance in order to calculate the proportions. To compute the Shannon diversity, you need the next column, labelled Ln(P), but to assist in the variance calculations it is convenient to calculate two others, which you can see in the table.
Using the spreadsheet for calculations
The spreadsheet Shannon diversity t-test calculator.xlsx will carry out the calculations for you. There are worksheets for two samples. You can enter a site name and your abundance values as well as species names if you like. The spreadsheet is protected (although there is no password) but you can add additional rows to accommodate extra data (row 23 is empty and that’s where you can start adding).
I advise that you don’t remove the worksheet protection, as it is easy to inadvertently change a formula! I’ve designed the spreadsheet so you can add extra rows without needing to remove the protection.
Enter the data for the two samples and the final t-test results will appear in the Results worksheet.
Calculating degrees of freedom
Once you have a value for t you need to determine if it is statistically significant. In order to do that you’ll need to work out the degrees of freedom. This is computed using the following formula.

Formula to calculate the degrees of freedom in the Hutcheson t-test.
In the formula you need the variance for each sample and the total abundance for each sample. The final value is close to the total abundance for the two samples added together.
Assessing statistical significance
Once you have your value for t and you have the appropriate degrees of freedom you can determine if the result is statistically significant. You can use the degrees of freedom to look up a critical value. If your calculated t-value exceeds the critical then your result is significant.
You can use Excel to calculate a critical value using the =TINV function. You need the level of significance (usually 0.05) and the degrees of freedom.
You can also determine the probability directly using the =TDIST function. You need your calculated t-value and the degrees of freedom.
The Shannon diversity t-test calculator.xlsx spreadsheet will display all the t-test results in the Results worksheet. The spreadsheet will also calculate confidence intervals and produce a couple of graphs (which you can edit).
Computing confidence intervals
It is useful to be able to calculate confidence intervals to your results. The confidence intervals allow you to compare multiple samples and to visualize how “different” samples are to one another.
You can compute a reasonable confidence interval using the standard deviation of the Shannon index (that is the square root of the variance) and multiplying by 2. With most communities you will have fairly large degrees of freedom and the critical value (for t) will approach 2 (the critical value for t at infinity is 1.96).
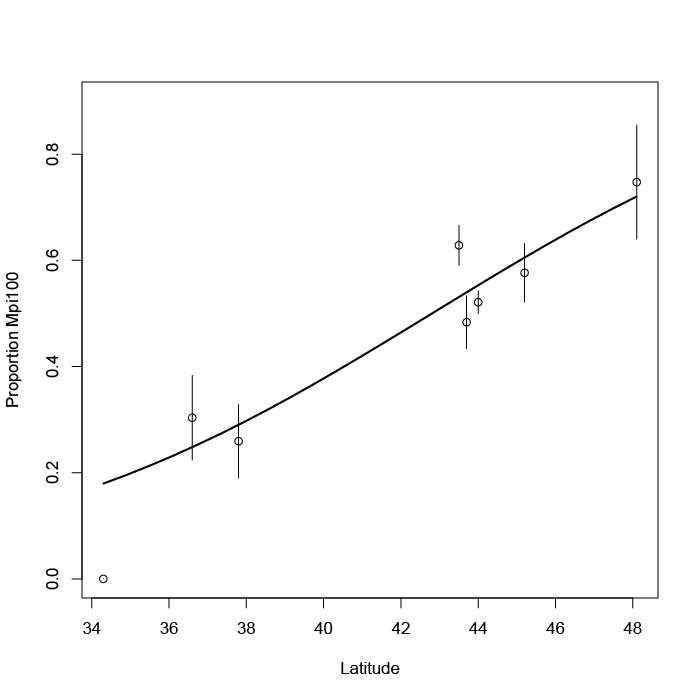
Once you have your confidence interval(s) you can add them to a graph to show the variability and possible overlap between samples. The Shannon diversity t-test calculator.xlsx will produce two charts (a bar chart and a point chart) with error bars based on the confidence intervals. You can edit the charts.
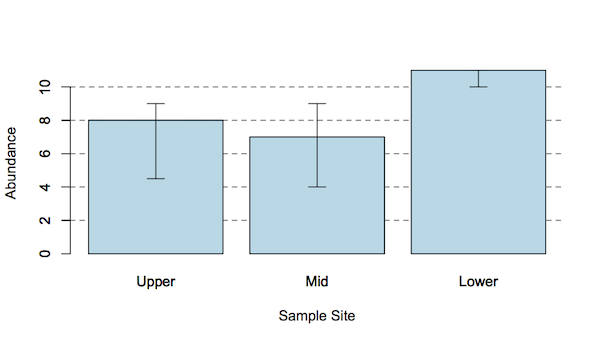
Graphing the results
It is useful to chart your results, the Shannon diversity t-test calculator.xlsx will produce two graphs, which you can edit. The error bars are based on the confidence interval.

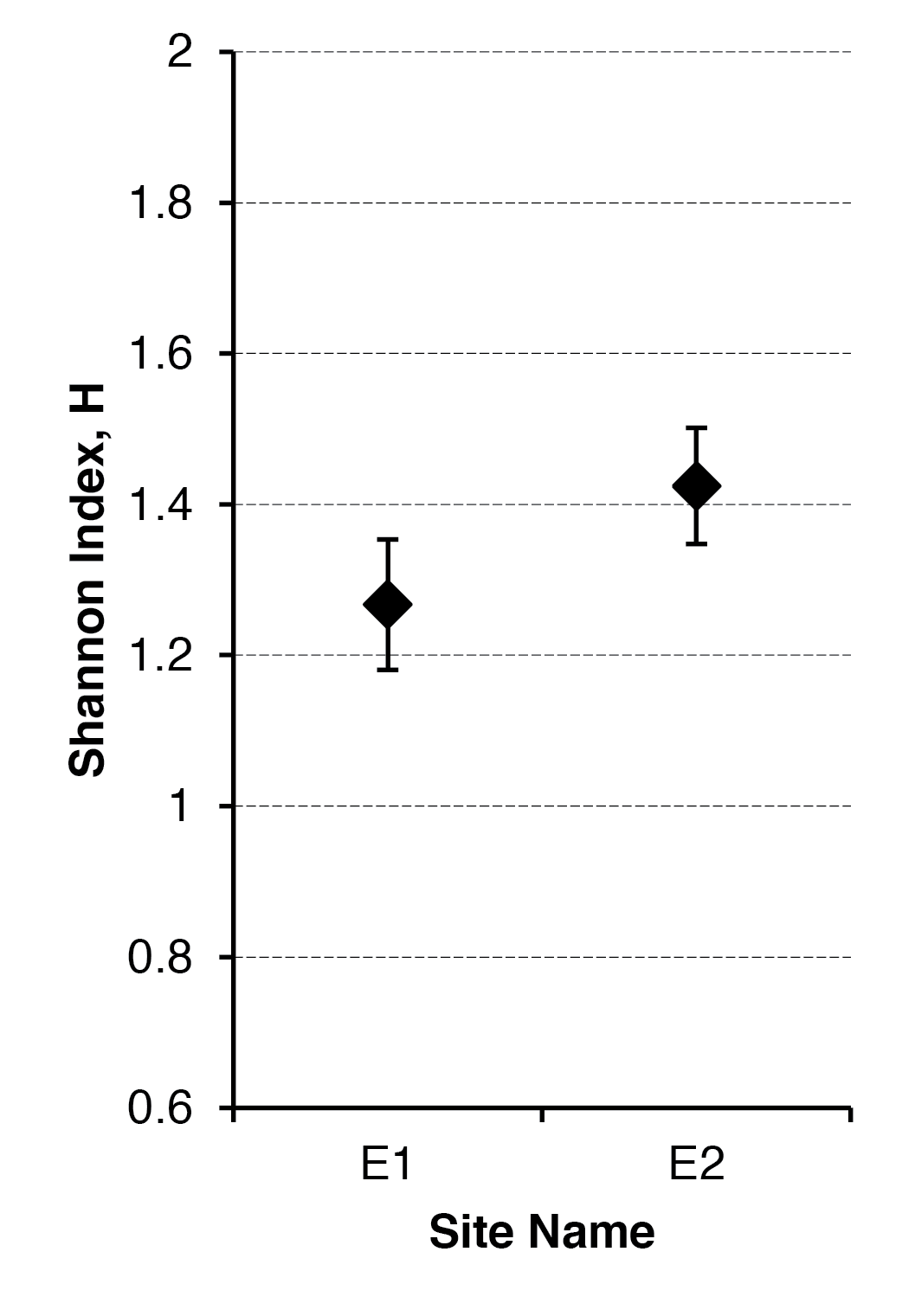
Two ways to represent the difference between Shannon diversity. Error bars are 95% confidence intervals.
The bar chart is a “classic” form of graph to show differences between samples. The point chart is a good alternative, as it allows you to see difference between samples more easily, especially if you customize the y-axis scale. The chart shown here would benefit from having the axis run from 1.0 to 1.6 for example.