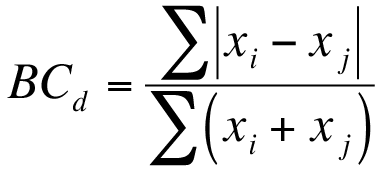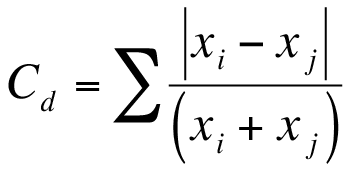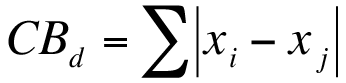The analysis of variance is a commonly used method to determine differences between several samples. R provides a method to conduct ANOVA via the aov() function. Your data need to be arranged in a particular manner for the aov() command to “work” (see 13). Your data need to be set out so that each column represents a single variable like so:
> head(fly) size diet 1 75 C 2 67 C 3 70 C 4 75 C 5 65 C 6 71 C
In this example the first column (size) is the response variable (dependent variable) and the second column (diet) is the predictor (independent) variable. ANOVA is a method of comparing samples that are grouped. The simplest arrangement is where you have a single grouping variable (predictor); this is called one-way ANOVA. If you have two predictor variables the analysis is called two-way ANOVA and so on.
The aov() command requires a formula that describes the experimental setup. In general terms you have:
response ~ predictor
This describes a 1-way anova.
ANOVA One-way
In one-way anova you have a response (dependent) variable and one predictor (grouping or independent) variable. It is usually best to assign a named object to “hold” the result of aov(), as there are multiple components you can make use of.
> mod = aov(size ~ diet, data = fly)
> summary(mod)
Df Sum Sq Mean Sq F value Pr(>F)
diet 4 1077.3 269.33 49.37 6.74e-16 ***
Residuals 45 245.5 5.46
—
Signif. codes: 0 ‘***’ 0.001 ‘**’ 0.01 ‘*’ 0.05 ‘.’ 0.1 ‘ ’ 1
The summary() command produces an anova table, a standard way to present such results. The table gives all the information you require.
Post hoc testing
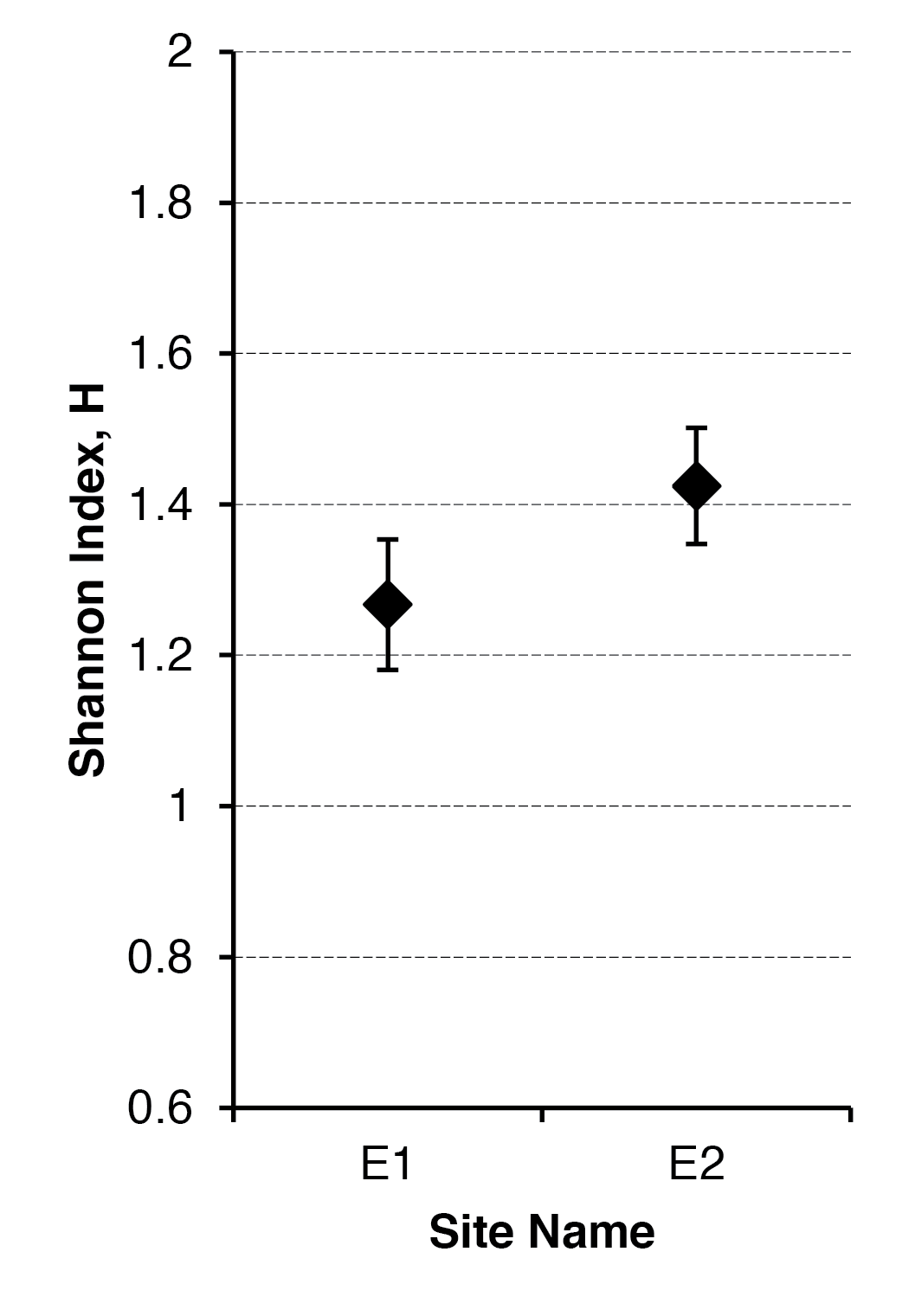
The basic ANOVA produces an overall result. In the previous example you can see a significant result; the different diets produce different sizes of fly wings! However, this is not the complete story, are all the groups different from one another or are some different and some not? What you need is a post hoc test. This carries out pair by pair analyses and takes into account the fact that you are running multiple tests (and so more likely to get a significant result by chance).
There are a number of different methods but the Tukey HSD test is the most common, and the TukeyHSD() command is built-in to R. You simply run the command on the result of aov(). In this case I used ordered = TRUE to make the results more readable:
> TukeyHSD(mod, ordered = TRUE)
Tukey multiple comparisons of means
95% family-wise confidence level
factor levels have been ordered
Fit: aov(formula = size ~ diet, data = fly)
$diet
diff lwr upr p adj
F-F.G 0.2 -2.768072 3.168072 0.9996878
G-F.G 1.3 -1.668072 4.268072 0.7256157
S-F.G 6.1 3.131928 9.068072 0.0000052
C-F.G 12.1 9.131928 15.068072 0.0000000
G-F 1.1 -1.868072 4.068072 0.8291029
S-F 5.9 2.931928 8.868072 0.0000100
C-F 11.9 8.931928 14.868072 0.0000000
S-G 4.8 1.831928 7.768072 0.0003242
C-G 10.8 7.831928 13.768072 0.0000000
C-S 6.0 3.031928 8.968072 0.0000072
The ordered = TRUE parameter makes sure that the pairs are compared in an order that ensures that the differences in means are always positive. So for example, F-C gives 11.9 as a mean difference but C-F would be -11.9. This simply makes the result easier to read.
the result shows the difference in mean value as well as 95% confidence intervals. The final column gives an adjusted p-value (the significance). You can see that some of the comparisons are not significant (values > 0.05), whilst others are (values < 0.05).
ANOVA models
The aov() command uses a formula to describe the setup. The formula is a powerful syntax that allows you to specify complicated anova models. Here are just a few examples:
| Model | Meaning |
| y ~ a | A one-way anova where a is a predictor (grouping variable). |
| y ~ a + b | A two-way anova where a and b are different predictors acting independently. |
| y ~ a * b | A two-way anova with a and b as predictors but where there is also an interaction between them. |
| y ~ a + b + a:b | The same as above but with the interaction written explicitly. |
| y ~ a + b + Error(c) | A two-way anova with a and b as predictors. Another variable, c, forms the error term, that is the within group variation. This is the way to describe repeated measures anova models. |
| y ~ a * b + Error(c/b) | This is a two-way repeated measures anova where b is nested in c and the predictors a and b have interaction. |
So, the formula syntax can describe quite complicated situations! The preceding table shows but a few of the options.